Chipster can use raw reads in fastq format, aligned reads in BAM format, variants in VCF format, and read count tables. You can also import reference genomes in fasta format and annotations in GTF and BED format. Fastq, fasta and GTF files can be compressed (.gz). In order to import these files to Chipster, select File / Import files in the top panel. File type specific importing instructions can be found below.
BAM and SAM filesSAM files need to be converted to BAM, and BAM files need to be sorted by chromosomal location and indexed. You will be prompted to preprocess the files during the import as shown in the screenshot below. This preprocessing runs on your own computer and it can take time depending on the file size. If you prefer, you can also run the sorting and indexing on the server: skip preprocessing, import the file, and run the tool "Utilities / Sort and index BAM" (or "Convert SAM to BAM, sort and index" if you have a SAM file). Note that if your data is already in the right format and your BAM file has the matching index file (.bai), you can use your data in Chipster without any preprocessing.
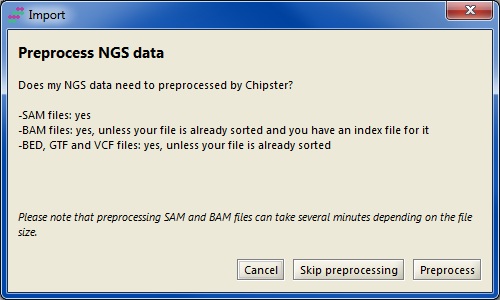
These files need to be sorted by chromosomal location. Like for BAM files, you can perform the preprocessing during data import (see above), or you can use sorting tools for these different file types in the Utilities category.
Read count tableRead count table should contain genes as rows and samples as columns, and it should be a tab delimited text file (save your table as text in Excel). When you select "File / Import files", you are prompted to choose an action to your file. Using the pull-down menu, select "Use Import tool" and click OK. Your file will now open in the Import tool, where you can define which rows and columns you want to use (see the Import tool manual for detailed instructions). Typically in step 1 you mark the title row, and in step 2 you mark the identifier column as Identifier and the count columns as Sample. When you click Finish, the samples will be imported as separate files to Chipster. Select all of them, and run the tool Utilities / Preprocess count table. This will create two files: a count table in Chipster format, and a phenodata file where you can describe your experimental setup. Use numbers to describe the experimental groups in the group column, so that the control group gets a smaller number.