Automating your data analysis with workflows
Data analysis typically involves several analysis steps.
Once a suitable combination has been found and analysis completed, it is often desirable to save the steps taken as an automatic workflow.
Reusing workflows serves many purposes. Firstly, it saves time as multi-step analysis can be executed with just one mouse click.
Sharing workflows within a research group brings consistency to analysis and provides an easy way for bioinformaticians to help biologists.
Sharing workflows in a wider context is also beneficial as providing a downloadable workflow file facilitates the reproduction of published results.
In addition to supporting user-made workflows, Chipster also includes some ready-made workflows for finding and analyzing differentially expressed genes, miRNAs and proteins.
User-made workflows
Chipster keeps track of the analysis steps taken, and displays them visually in the Workflow-panel.
You can experiment with different methods and parameters, and prune the resulting workflow by simply deleting the unwanted steps.
When a satisfactory analysis pipeline is ready, you simply click on the beginning point of it and save the workflow by selecting Workflow->Save starting from selected. You can save the workflow file anywhere you like and change its name, but the ending has to be .bsh.
The workflow is saved as a file, which contains instructions to run certain analysis tools with the selected parameter settings in a certain order.
Note that you can also run a workflow for several identical input files such as FASTQ or BAM at the same time: Simply select all the input files, and the option "Run recent for each".
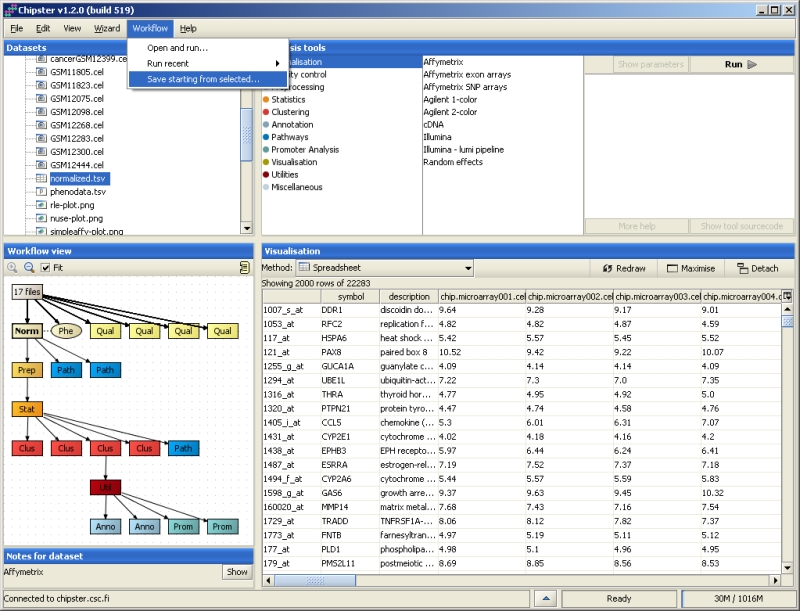
If you would like to apply the same workflow to another normalized dataset, select Workflow->Open and run or Workflow->Run recent. You can also share a workflow with others by simply giving them a copy of your workflow file.
Ready-made workflows
Chipster's ready-made workflows are started from the menu Workflows / Run from repository. They cover finding and analyzing differentially expressed genes, miRNAs and proteins as described below.
Please note that these workflows should be run on normalized data and
the experimental groups needs to be defined in the phenodata.
1. Find differentially expressed genes
This workflow performs quality control of the samples, finds differentially expressed genes, and runs annotation and clustering from them.
- Quality control of samples: NMDS, PCA (view the results with 3D scatter plot) and dendrogram.
- Filtering out non-changing genes: Filter by standard deviation (filters out 99.7% of the genes)
- Statistical test to find differentially expressed genes: Two group test using EmpiricalBayes, p-value < 0.05, p-value adjustment by BH method.
- Annotation
- Sort samples according to group
- Clustering: Hierarchical clustering using Pearson correlation as distance method and average linkage for drawing the dendrogram.
2. Find differentially expressed miRNAs
This workflow performs quality control of the samples, finds differentially expressed miRNAs, and runs annotation and clustering from them.
It also performs pathway analysis using KEGG and GO databases for the potential targets of these miRNAs. Please use
the miRNA systematic names (e.g. hsa-miR-100) as identifiers when importing your data to Chipster. Note that this workflow
takes time to run because of the large pathway analysis part.
- Quality control of samples: NMDS, PCA (view the results with 3D scatter plot) and dendrogram.
- Filtering out non-changing miRNAs: Filter by coefficient of variation (filters out 50% of the miRNAs)
- Statistical test to find differentially expressed genes: Two group test using EmpiricalBayes, p-value < 0.01, p-value adjustment by BH method.
- Annotation
- Pathway analysis: Finds putative target genes for the differentially expressed miRNAs from miRBase
and performs a hypergeometric test for their enrichment in GO categories and KEGG pathways.
- Clustering: Hierarchical clustering using Pearson correlation as distance method and average linkage for drawing the dendrogram.
- Filter for fold change: Find those differentially expressed miRNAs which are 2-fold up- or downregulated.
3. Find differentially expressed proteins
This workflow performs quality control of the samples, finds differentially expressed proteins, and runs pathway analysis and clustering for them.
Please use UniProt accession numbers (e.g. Q5STU3) as identifiers when importing your data to Chipster. Marking also an annotation column in the Import tool allows
Chipster to show these annotations in visualizations. Please note that if your data is not log-transformed, you can transform it in Chipster prior to running the workflow
using the tool "Utilities/ Change interpretation".
- Missing values: Imputation with the KNN method using 5 neighbors.
- Normalization to chip median.
- Quality control of samples: Boxplot, histogram, PCA (view the results with 3D scatter plot) and dendrogram.
- Filtering out non-changing proteins: Filter by coefficient of variation (filters out 50% of the proteins)
- Statistical test to find differentially expressed proteins: Two group test using EmpiricalBayes, p-value < 0.05, p-value adjustment by BH method.
- Protein-protein interactions: Interaction partners for the differentially expressed proteins are retrieved from the IntAct database.
- Mapping to pathways: Differentially expressed proteins are mapped to Reactome pathways.
- Pathway enrichment: Hypergeometric test using the ConsensusPathDB.
- Sort samples according to group
- Clustering: Hierarchical clustering using Pearson correlation as distance method and average linkage for drawing the dendrogram.