Directional/stranded RNA-seq data -which parameters to choose?
"Directional" or "stranded" RNA-seq methods are gaining popularity. Several protocols and products are available
for the library preparation step, and different tools and softwares have different options to take
these into account. Since this has caused a lot of confusion due to incoherent parameter naming,
we try to clarify this issue a bit here.
To be able to select the right parameters for your data, first you need to know which library prep
method was used when generating your data. In general, there are three types of library preps:
- unstranded
- second-strand = directional, where the first read of the read pair
(or in case of single end reads, the only read) is from the transcript strand
- first-strand = directional, where the first read (or the only read in case of SE) is from the opposite
strand.
Summary of library type protocols
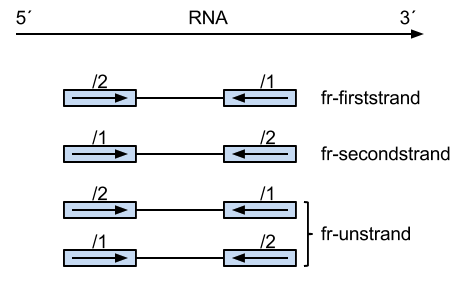
(Image borrowed from:
http://onetipperday.sterding.com/2012/07/how-to-tell-which-library-type-to-use.html)
The reads on the left are from the same strand as the transcript, and their pairs on the right are from the
opposing strand.
The number above the read states which read it is, the first (/1) or the second (/2). Thus, perhaps a bit
unintuitively, the first case,
"fr-firststrand" is such that the first read (/1) is actually from the opposing strand as the transcript, and second
read (/2) is from the transcript strand.
Why is this so important?
- If you use wrong directionality parameter in the read counting step with HTSeq,
the reads are considered to be from the wrong strand. This means that in the case where there
is no gene on that other strand, you won't get any counts, and if there is a gene in the same
location on the other strand, your reads are counted for the wrong gene.
- If you use wrong directionality parameter in the reference alignment step, the XS tag in the resulting BAM file will contain wrong strand information.
The XS tag is used by transcript assembly programs like Cufflinks and Stringtie, and also Cuffdiff uses it.
How can I check that I chose correctly?
It's a good idea to check that!
Be extra careful to assign the paired files correctly! Using these parameters assumes you are giving the
files in specific order: read1, read2. In Chipster always check from the parameters window that your
files are assigned correctly.
Below we list some common library preparation kits and their corresponding parameters
in different tools. Is your kit missing from the list? If you have the data generated with that kit and
figure
out the library type, please let us know too, so we can add that kit to the list below.
Unstranded:
Information regarding the strand is not conserved
(it is lost during the amplification of the mRNA fragments).
Kits:
TruSeq RNA Sample Prep kit
Parameters:
TopHat / Cufflinks / Cuffdiff: library-type fr-unstranded
HISAT2: default
HTSeq: stranded -- no
Directional, first strand:
The second read (read 2) is from the original RNA strand/template, first read (read 1) is from the
opposite strand.
The information of the strand is preserved as the original RNA strand is degradated due to the
dUTPs incorporated in the second synthesis step.
Kits:
All dUTP methods, NSR, NNSR
TruSeq Stranded Total RNA Sample Prep Kit
TruSeq Stranded mRNA Sample Prep Kit
NEB Ultra Directional RNA Library Prep Kit
Agilent SureSelect Strand-Specific
Parameters:
TopHat / Cufflinks / Cuffdiff: library-type fr-firststrand
HISAT2: --rna-strandedness R (for SE) / RF (for PE)
HTSeq: stranded -- reverse
Directional, second strand:
The first read (read 1) is from the original RNA strand/template, second read (read 2) is from the
opposite strand.
The directionality is preserved, as different adapters are ligated to different ends of the fragment.
Kits:
Directional Illumina (Ligation), Standard SOLiD
ScriptSeq v2 RNA-Seq Library Preparation Kit
SMARTer Stranded Total RNA
Encore Complete RNA-Seq Library Systems
NuGEN SoLo
Parameters:
TopHat / Cufflinks / Cuffdiff: library-type fr-secondstrand
HISAT2: --rna-strandedness F (for SE) / FR (for PE)
HTSeq: stranded -- yes
Summary of parameters:
| Tool |
Unstranded |
Read 1 and transcript on the same strand |
Read 1 on the opposite strand |
| RSeQc |
- |
++,-- (SE) 1++,1--,2+-,2-+ (PE) |
+-,-+ (SE) 1+-,1-+,2++,2-- (PE) |
| TopHat / Cufflinks |
library-type fr-unstranded |
library-type fr-secondstrand |
library-type fr-firststrand |
| HISAT2 |
default |
--rna-strandedness F (SE), FR (PE) |
--rna-strandedness R (SE), RF (PE) |
| HTSeq |
stranded --no |
stranded --yes |
stranded --reverse) |
Don't get confused: Bowtie parameters
Note also that the --fr/--rf/--ff or "Order of mates to align" parameter in Bowtie has similar
sounding parameter options: [--fr: "Forward/reverse", --rf: "Reverse/Forward", --ff: "Forward/forward"].
However, these parameters are a bit different story, as they explain how the paired end reads are
oriented towards each other (-> <-, -> -> or <- ->). The default (--fr, -> <-) is appropriate for Illumina's
paired-end reads: it means that read 1 appears upstream of the reverse complement of read 2, or vice
versa. When running TopHat, the library-type parameter is delivered to Bowtie, so the user doesn't
have to worry about that too much.