This tool uses Bowtie2 software to align paired-end reads to a reference genome or sequence set. You need to supply the reads in two or more files containing the reads in the same order and a FASTA formatted reference sequence. Reads can be in either FASTA or FASTQ format, but all reads files need to be in the same format.
After running Bowtie2, Chipster converts the alignment file to BAM format, and sorts and indexes it using the SAMtools package.
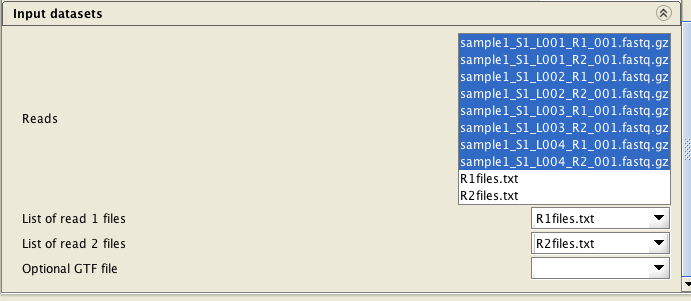
If you have more than two reads files, you will need to provide a list of filenames of the reads files for each direction (one file for read1 files, and another one for the read2 files) as a text file (e.g.R1files.txt and R2files.txt). These lists can be generated with the tool Utilities / Make a list of file names . The read pairs must be ordered identically in both lists.
Select BOTH list files (R1files.txt and R2files.txt), ALL reads files (8 of them in the example below) and the
reference FASTA, and
assign the list files correctly. When assigning the list files, they are automatically inactivated in the
"reads" file list.
Bowtie2 uses heuristics for mapping the reads to the reference genome. This means that the reported mapping is not guaranteed to be the best mapping. Several Bowtie2 parameters affect to the mapping accuracy. However higher accuracy (sensitivity) also requires more computing time. In Chipster you can choose, for both end-to-end and local alignment strategy, the sensitivity level from a set of pre-defined parameter combinations that allow you to tune the balance between the computing time and mapping sensitivity:
| Strategy | Corresponding command line options |
|---|---|
| Very fast | -D 5 -R 1 -N 0 -L 22 -i S,0,2.50 |
| Fast | -D 10 -R 2 -N 0 -L 22 -i S,0,2.50 |
| Sensitive (default) | -D 15 -R 2 -N 0 -L 22 -i S,1,1.15 |
| Very sensitive | -D 20 -R 3 -N 0 -L 20 -i S,1,0.50 |
| Very fast local | --local -D 5 -R 1 -N 0 -L 25 -i S,1,2.00 |
| Fast local | --local -D 10 -R 2 -N 0 -L 22 -i S,1,1.75 |
| Sensitive local | --local -D 15 -R 2 -N 0 -L 20 -i S,1,0.75 |
| Very sensitive local | --local -D 20 -R 3 -N 0 -L 20 -i S,1,0.50 |
By default Chipster does the alignment using the end-to-end alignment method with "Sensitive" parameters.
In addition to the mapping strategy, the Bowtie2 tool in Chipster allows users to modify many other mapping and output parameters too. Please check the Bowtie2 manual for more detailed description of these parameters.
| Parameter | Corresponding command line option | Description of the parameter |
|---|---|---|
| Quality value format used | --phred33, --phred64 or --ignore-quals | Quality scale used in the fastq-file. |
| How many valid alignments are reported per read | none, -k or --all | By default, Bowtie2 reports only the best alignment of the read (based on the mapping quality\). Optionally, if there are several, equally good alignments, you can choose how many of them should be reported? |
| Put reads that did not align concordantly to a separate file | --un-conc | Write paired-end reads that fail to align concordantly to a fastq file. |
| Match bonus | --ma | Match bonus for a match in local alignment. Default value 2 |
| Maximum penalty for mismatch | --mp | Maximum penalty for mismatch; lower quality = lower penalty. Default value 6 |
| Penalty for non-ACGTs | --np | Sets penalty for positions where the read, reference, or both, contain an ambiguous character such as N. Default: 1. |
| Gap opening penalty for the reads | --rdg | Gap opening penalty for the reads. Default value: 5. |
| Gap extension penalty for the reads | --rdg | Gap extension penalty for the reads. Default value: 3. |
| Gap opening penalty for the reference | --rfg | Gap opening penalty for the reference. Default value: 5. |
| Gap extension penalty for the reference | --rfg | Gap extension penalty for the reference. Default value: 3. |
| Minimum insert length | --minins | Minimum insert length between the mate pairs. Default value: 0 |
| Maximum insert length | --maxins | Maximum insert length between the mate pairs. Default value: 500 |
| Order of mates to align | --fr, --rf or --ff | The orientation of the mate pairs. Default: forward/revrse |
| Suppress unpaired alignments | --no-mixed | By default, when bowtie2 cannot find a concordant or discordant alignment for a pair, it then tries to find alignments for the individual mates. This option disables that behavior. |
| Suppress discordant alignments | --no-discordant | By default, bowtie2 looks for discordant alignments if it cannot find any concordant alignments. A discordant alignment is an alignment where both mates align uniquely, but that does not satisfy the paired-end constraints. This option disables that behavior |
| Not concordant when mates extend past each other | --no-dovetail | If the mates "dovetail", that is if one mate alignment extends past the beginning of the other such that the wrong mate begins upstream, consider that to be concordant. Default: mates cannot dovetail in a concordant alignment. |
| Not concordant when one mate alignment contains other | --no-contain | If one mate alignment contains the other, consider that to be non-concordant. Default: a mate can contain the other in a concordant alignment. |
| Not concordant when mates overlap at all | --no-overlap | If one mate alignment overlaps the other at all, consider that to be non-concordant. Default: mates can overlap in a concordant alignment. |
This tool returns the alignment in BAM format and an index file for it (.bai). It also produces a log file, which allows you to see what percentage of the reads align with the selected parameter settings. Optionally also fastq files are produced for the unaligned reads, or reads that align discordantly.