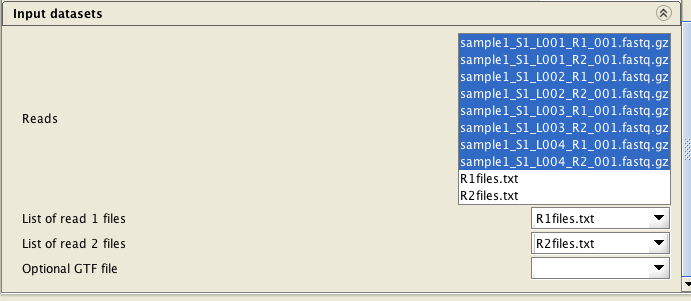
Select BOTH list files (R1files.txt and R2files.txt) and ALL FASTQ files (8 of them in the example below), and assign the list files correctly. When assigning the list files, they are automatically inactivated in the "reads" file list.
Aligns paired end Illumina RNA-seq reads to a genome provided as a FASTA file in order to identify exon-exon splice junctions.
You need to supply the reads in FASTQ files. The files can be compressed with gzip. Note that if you have more than two FASTQ files per sample (for example, Illumina NextSeq produces 8 FASTQ files per sample), you need to provide also two list files containing the file names in order to assign the FASTQ files to each direction. Please produce the list files using the tool "Utilities / Make a list of file names".
List files are optional if you provide just two FASTQ files. Chipster will try to assign the files to directions based on file names. This assumes the files are named so that the beginning of the name is identical and the directions are specified with _1 and _2, e.g. Abc123_1, Abc123_2. If your files are named differently, you need to provide list files to make sure the files are assigned correctly.
If you have more than two FASTQ files, you will need to provide a list of filenames of the FASTQ files for each direction as a text file (e.g.1files.txt and 2files.txt). These lists can be generated with the tool Utilities / Make a list of file names. The read pairs must be ordered identically in both lists.
Supplying a GTF file containing known gene and exon locations is recommended, because it improves the alignment process.
TopHat2 maps Illumina RNA-Seq reads to a genome in order to identify exon-exon splice junctions. The alignment process consists of several steps. If annotation is available as a GTF file, TopHat will extract the transcript sequences and use Bowtie2 to align reads to this virtual transcriptome first. Only the reads that do not fully map to the transcriptome will then be mapped on the genome. The reads that still remain unmapped are split into shorter segments, which are then aligned to the genome. Segment mappings are used to find potential splice sites. Sequences flanking a splice site are concatenated, and unmapped segments are mapped to them. Segment alignments are then stitched together to form whole read alignments.
The "anchor length" means that TopHat2 will report junctions spanned by reads with at least this many bases on each side of the junction. Note that individual spliced alignments may span a junction with fewer than this many bases on one side. However, every junction involved in spliced alignments is supported by at least one read with this many bases on each side. By default no mismatches are allowed in the anchor, but you can change this.
TopHat2 will ignore donor-acceptor pairs which are closer than the minimum intron length or further than the maximum intron length apart.
For paired-end reads, TopHat2 processes the two reads separately through the same mapping stages described above. In the final stage, the independently aligned reads are analyzed together to produce paired alignments, taking into consideration additional factors including fragment length and orientation. The expected mean inner distance between mate pairs means the fragment length minus the reads. For example, if your fragment size is 300 bp and read length is 50 bp, the inner distance is 200.
If you have more than two FASTQ files, you will need to provide a list of filenames of the FASTQ files for each direction (one file for read1 files, and another one for the read2 files) as a text
file (e.g.R1files.txt and R2files.txt). These lists can be generated with the tool Utilities / Make a list of file names . The read
pairs must be ordered identically in both lists.
Select BOTH list files (R1files.txt and R2files.txt) and ALL FASTQ files (8 of them in the example below), and assign the list files correctly.
When assigning the list files, they are automatically inactivated in the "reads" file list.
If your RNA-seq data was produced with a stranded/directional protocol, it is important that you select the correct strandedness option in the parameter "Library type":
After running TopHat2, Chipster indexes the BAM file using the SAMtools package. This way the results are ready to be visualized in the genome browser.
This tool returns the following files:
This tool is based on the TopHat package. Please cite the following article:
Kim D, Petrtea G, Trapnell C, et al. TopHat2: accurate alignments of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology (2013) 14: R36.